
Genetic Variation and Phylogeography of Lumbriculus variegatus (Annelida: Clitellata: Lumbriculidae) Based on Mitochondrial Genes
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimens Collection, DNA Extraction and PCR Amplification
2.2. Population Genetic Analysis
2.3. Phylogenetic Analysis
2.4. Haplotype Analysis
2.5. Biogeographic Analysis
3. Results
3.1. Sequence Divergence Analysis
3.2. Distance and Population Structure Analysis
3.3. Phylogenetic Relationships
3.4. Haplotype Analysis
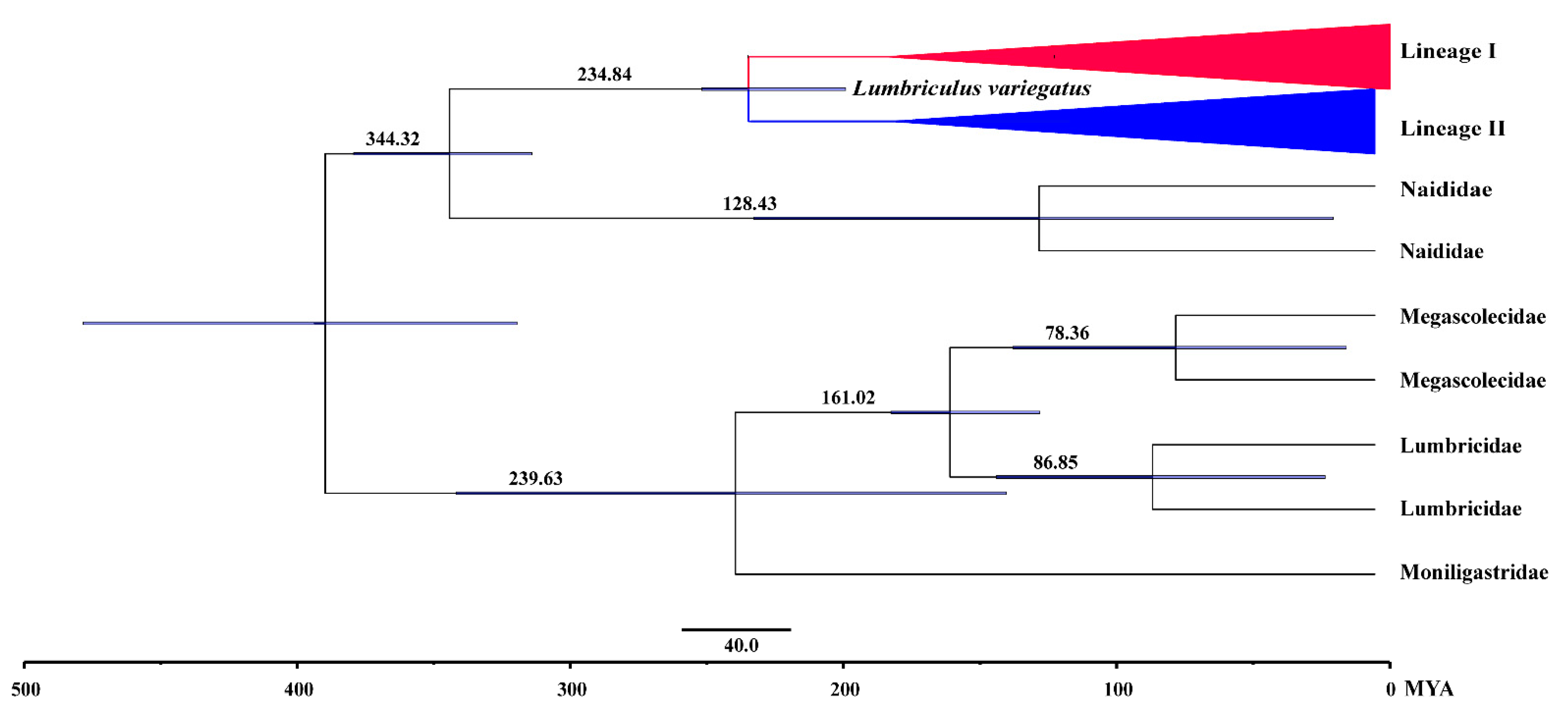
3.5. Biogeographic Analysis
4. Discussion
4.1. Genetic Variation
4.2. Cryptic Species
4.3. Population Historical Dynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Müller, O.F. Vermium Terrestrium et Fluviatilium; Havniae et Lipsiae: Leipzig, German, 1774; pp. 1773–1774. [Google Scholar]
- Claparède, E. Recherches sur les Oligochètes. In Memoires de la Société de Physique et D’histoire Naturelle de Genève; Société de Physique et d’Histoire Naturelle de Genève: Geneva, Switzerland, 1862; Volume 16, p. 217. [Google Scholar]
- Lumbriculus variegatus (Müller, 1774) in GBIF Secretariat. GBIF Backbone Taxonomy. Checklist Dataset. Available online: https://doi.org/10.15468/39omei (accessed on 17 November 2022).
- Chapman, P.M. Introduction to perspectives: Aquatic behavioural ecotoxicology—Coming of Age. Hum. Ecol. Risk Assess. 2007, 13, 478–480. [Google Scholar] [CrossRef]
- Gustafsson, D.R.; Price, D.A.; Erséus, C. Genetic variation in the popular lab worm Lumbriculus variegatus (Annelida: Clitellata: Lumbriculidae) reveals cryptic speciation. Mol. Phylogenet. Evol. 2009, 51, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Kontchou, J.A.; Nachev, M.; Sures, B. Ecotoxicological effects of traffic-related metal sediment pollution in Lumbriculus variegatus and Gammarus sp. Environ. Pollut. 2021, 268 Pt B, 115884. [Google Scholar] [CrossRef]
- Marchan, D.F.; Fernandez, R.; Sosa, I.D.; Cosin, D.; Novo, M. Pinpointing cryptic borders: Fine-scale phylogeography and genetic landscape analysis of the Hormogaster elisae complex (Oligochaeta, Hormogastridae). Mol. Phylogenet. Evol. 2017, 112, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Aspe, N.M.; James, S.W. Molecular phylogeny and biogeographic distribution of pheretimoid earthworms (Clitellata: Megascolecidae) of the Philippine archipelago. Eur. J. Soil Biol. 2018, 85, 89–97. [Google Scholar] [CrossRef]
- Shen, H.P.; Chang, C.H.; Ota, H. The biogeographical history of giant earthworms of the Metaphire formosae species group (Clitellata: Megascolecidae) in Taiwan and the Ryukyu Archipelago, with the description of a new species from Yonagunijima, Southern Ryukyus. Org. Divers. Evol. 2022, 22, 47–60. [Google Scholar] [CrossRef]
- Erséus, C.; Williams, B.W.; Horn, K.M.; Halanych, K.M.; Santos, S.R.; James, S.W.; Châtelliers, M.C.; Anderson, F.E. Phylogenomic analyses reveal a Palaeozoic radiation and support a freshwater origin for clitellate annelids. Zool. Scr. 2020, 49, 614–640. [Google Scholar] [CrossRef]
- Mcgaugh, S.E.; Eckerman, C.M.; Janzen, F.J. Molecular phylogeography of Apalone spinifera (Reptilia, Trionychidae). Zool. Scr. 2008, 37, 289–304. [Google Scholar] [CrossRef]
- Sagonas, K.; Poulakakis, N.; Lymberakis, P.; Parmakelis, A.; Pafilis, P.; Valakos, E.D. Molecular systematics and historical biogeography of the green lizards (Lacerta) in Greece: Insights from mitochondrial and nuclear DNA. Mol. Phylogenet. Evol. 2014, 76, 144–154. [Google Scholar] [CrossRef]
- Sakai, H.; Ueda, T.; Yokoyama, R.; Safronov, S.N.; Goto, A. Genetic structure and phylogeography of northern Far East pond minnows, Rhynchocypris perenurus sachalinensis and R. p. mantschuricus (Pisces, Cyprinidae), inferred from mitochondrial DNA sequences. Biogeography 2014, 16, 87–109. [Google Scholar]
- Hou, Z.; Jin, P.; Liu, H.; Qiao, H.; Sket, B.; Cannizzaro, A.G.; Berg, D.J.; Li, S. Past climate cooling promoted global dispersal of amphipods from Tian Shan montane lakes to circumboreal lakes. Glob. Chang. Biol. 2022, 28, 3830–3845. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, A.; Ray, S.; Roy, A. Molecular markers in phylogenetic studies—A review. Phylogenet. Evol. Biol. 2014, 2. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; deWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folmer, O.; Black, M.B.; Hoeh, W.; Lutz, R.; Vrijenhoek, R.C. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Bely, A.E.; Wray, G.A. Molecular phylogeny of naidid worms (Annelida: Clitellata) based on cytochrome oxidase I. Mol. Phylogenet. Evol. 2004, 30, 50–63. [Google Scholar] [CrossRef]
- Palumbi, S.R.; Martin, A.; Romano, S.; McMillan, W.O.; Stice, L.; Grabowski, G. The Simple Fool’s Guide to PCR, Version 2.0; Palumbi, S., Ed.; Department of Zoology, University of Hawaii: Honolulu, HI, USA, 1991; p. 96822. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Tajima, F. The effect of change in population size on DNA polymorphism. Genetics 1989, 123, 597–601. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Haeseler, A.V.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. PopART: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Du, Y.Y.; Yang, Z.Y.; Zhang, Y.P.; Lou, Z.Y.; Jiao, W.L. Population genetic structure of Schizopygopsis kialingensis inferred from mitochondrial D-loop sequences. Acta Ecol. Sin. 2017, 37, 7741–7749. [Google Scholar] [CrossRef]
- Bonin, A.; Nicole, F.; François, P.; Miaud, C.; Taberlet, P. Population adaptive index: A new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv. Biol. 2007, 21, 697–708. [Google Scholar] [CrossRef]
- Hewitt, G.M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 1996, 58, 247–276. [Google Scholar] [CrossRef]
- Grant, W.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yang, Y.P.; Zhang, H.; Chen, Y.X. Genetic diversity and population demographic history of three populations of Barbatula toni (Cypriniformes, Nemacheilinae) from North China. Acta Hydrobiol. Sin. 2019, 43, 8. [Google Scholar] [CrossRef]
- Wright, S. The interpretation of population structure by f-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Liao, J.; Jing, D.; Luo, G.; Wang, Y.; Zhao, L.; Liu, N. Comparative phylogeography of Meriones meridianus, Dipus sagitta, and Allactaga sibirica: Potential indicators of the impact of the Qinghai-Tibetan plateau uplift. Mamm. Biol. 2016, 81, 31–39. [Google Scholar] [CrossRef]
- Crottini, A.; Marotta, R.; Barbuto, M.; Casiraghi, M.; Ferraguti, M. The world in a river? A preliminary analysis of the 16S rDNA variability of Tubifex species (Clitellata: Tubificidae) from the Lambro River. Mol. Phylogenetics Evol. 2008, 48, 1189–1203. [Google Scholar] [CrossRef] [PubMed]
- Vivien, R.; Holzmann, M.; Werner, I.; Pawlowski, J.; Lafont, M.; Ferrari, B.D. Cytochrome c oxidase barcodes for aquatic oligochaete identification: Development of a Swiss reference database. PeerJ 2017, 5, e4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.K.; Fend, S.V.; Martinsson, S.; Erséus, C. Extensive cryptic diversity in the cosmopolitan sludge worm Limnodrilus hoffmeisteri (Clitellata, Naididae). Org. Divers. Evol. 2017, 17, 477–495. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.G. Family Lumbriculidae. In Aquatic Oligochaeta of the World; Brinkhurst, R.O., Jamieson, B.G.M., Eds.; Oliver and Boyd: Edinburgh, UK, 1971; pp. 200–285. [Google Scholar]
- Holmquist, C. Lumbriculids (Oligochaeta) of northern Alaska and northwestern Canada. Zool. Jb. Syst. 1976, 103, 377–431. [Google Scholar]
- Popchenko, V.I. Worms of the genus Lumbriculus Grube (Oligochaeta, Lumbriculidae) from water bodies of Karelia. Zool. Zhurnal 1976, 55, 1617–1626. [Google Scholar]
- Morev, A.P. New species of the family Lumbriculidae (Oligochaeta) from water bodies in the north-east of the USSR. Zool. Zhurnal 1982, 61, 663–670. [Google Scholar]
- Timm, T.; Rodriguez, P. Description of a new Lumbriculus species (Oligochaeta, Lumbriculidae) from the Russian Far-East. Ann. Limnol.—Int. J. Limnol. 1994, 30, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Smith, F. Notes on species of North American Oligochaeta. Bull. Ill. State Lab. Nat. Hist. 1895, 4, 285–297. [Google Scholar] [CrossRef]
- Yamaguchi, H. Studies on the aquatic Oligochaeta of Japan. I. Lumbriculids from Hokkaido. J. Fac. Sci. Hokkaido Univ. Zool. 1936, 5, 73–94. [Google Scholar]
- Sokolskaya, N.L. New species of the genus Lumbrículus Grube (Lumbriculidae, Oligochaeta) from South Sakhalin reservoirs. Byulleten Mosk. Obs. Ispyt. Prir. Otd. Biol. 1967, 72, 40–47. [Google Scholar]
- Yamaguchi, H. Studies on the Aquatic Oligochaeta of Japan: Ⅵ. A Systematic Report, with Some Remarks on the Classification and Phylogeny of the Oligochaeta (with 1 Plate, 5 Tables and 25 Text-figures). J. Fac. Sci. Hokkaido Univ. Ser. V. Zool. 1953, 11, 277–342. [Google Scholar]
- Yamaguchi, H. Studies on the Aquatic Oligochaeta of Japan: Ⅲ. A Description of Lumbriculus multiatriatus n. sp., with Remarks on Distribution of the Genital Organs in the Lumbriculidae. J. Fac. Sci. Hokkaido Imp. Univ. Ser. VI Zool. 1937, 6, 1–12. [Google Scholar]
- Sokolskaya, N.L. On the Lumbriculidae (Oligochaeta) fauna of the Chukchi Peninsula. Byulleten Mosk. Obs. Ispyt. Prir. Otd. Biol. 1976, 83, 43–53. [Google Scholar]
- Novo, M.; Almodóvar, A.; Fernández, R.; Giribet, G.; Díaz Cosín, D.J. Understanding the biogeography of a group of earth-worms in the Mediterranean basin—The phylogenetic puzzle of Hormogastridae (Clitellata: Oligochaeta). Mol. Phylogenet. Evol. 2011, 61, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Envall, I.; Gustavsson, L.M.; Erséus, C. Genetic and chaetal variation in Nais worms (Annelida, Clitellata, Naididae). Zool. J. Linn. Soc. 2012, 165, 495–520. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | Specimen Number | Haplotype Number | Haplotype Diversity (Hd) | Nucleotide Diversity (π) | Tajima’s D Test (D) | Tajima’s D Test (p) | Fu’s Fs Test (D) | Fu’s Fs Test (p) |
|---|---|---|---|---|---|---|---|---|
| I | 46 | 15 | 0.859 | 0.007 | −1.964 ** | 0.010 | −1.061 | 0.364 |
| II | 17 | 14 | 0.971 | 0.062 | 0.431 | 0.693 | 1.560 | 0.777 |
| Total | 63 | 29 | 0.923 | 0.062 | −0.767 | 0.352 | 0.249 | 0.571 |
| Lineage | I | II |
|---|---|---|
| I | 0.057 | 0.745 |
| II | 0.147 | 0.141 |
| Source of Variation | df | Sum of Squares | Variance Components | Percentage of Variation (%) |
|---|---|---|---|---|
| Among populations | 1 | 1021.652 | 40.809 | 82.65 |
| Within populations | 61 | 522.427 | 8.564 | 17.35 |
| Total | 62 | 1544.079 | 49.373 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Yu, J.; Zhao, Y.; He, D.; Wang, H.; Cui, Y. Genetic Variation and Phylogeography of Lumbriculus variegatus (Annelida: Clitellata: Lumbriculidae) Based on Mitochondrial Genes. Diversity 2023, 15, 158. https://doi.org/10.3390/d15020158
Zhou T, Yu J, Zhao Y, He D, Wang H, Cui Y. Genetic Variation and Phylogeography of Lumbriculus variegatus (Annelida: Clitellata: Lumbriculidae) Based on Mitochondrial Genes. Diversity. 2023; 15(2):158. https://doi.org/10.3390/d15020158
Chicago/Turabian StyleZhou, Tingting, Jiefeng Yu, Yongjing Zhao, Dekui He, Hongzhu Wang, and Yongde Cui. 2023. "Genetic Variation and Phylogeography of Lumbriculus variegatus (Annelida: Clitellata: Lumbriculidae) Based on Mitochondrial Genes" Diversity 15, no. 2: 158. https://doi.org/10.3390/d15020158