
A Novel GNAS Mutation in a Patient with Ia Pseudohypoparathyroidism (iPPSD2) Phenotype
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
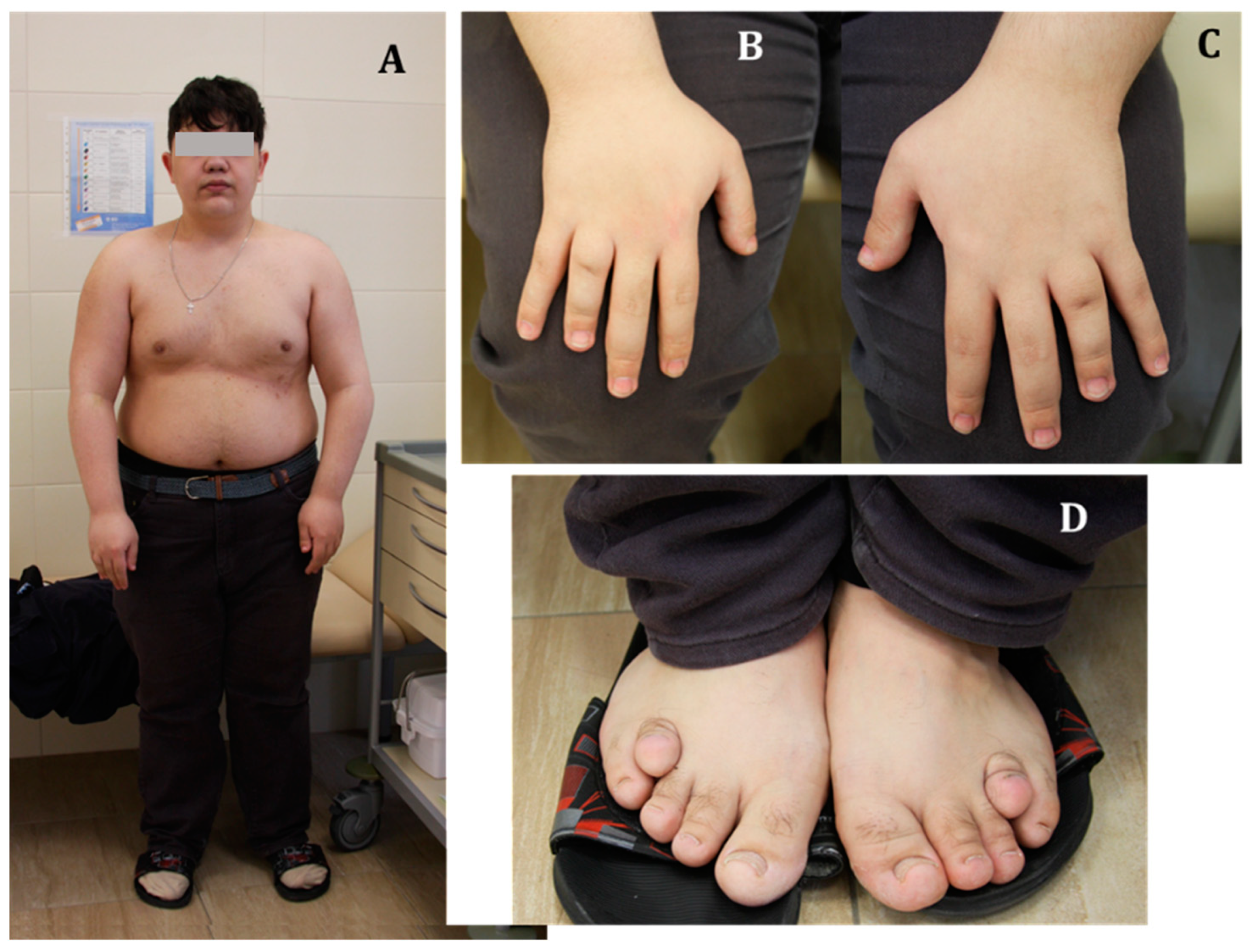
3.1. Case Description
3.2. Anamnesis Morbi
3.3. Physical Examination
3.4. Examination Data and Treatment
3.5. Follow-Up
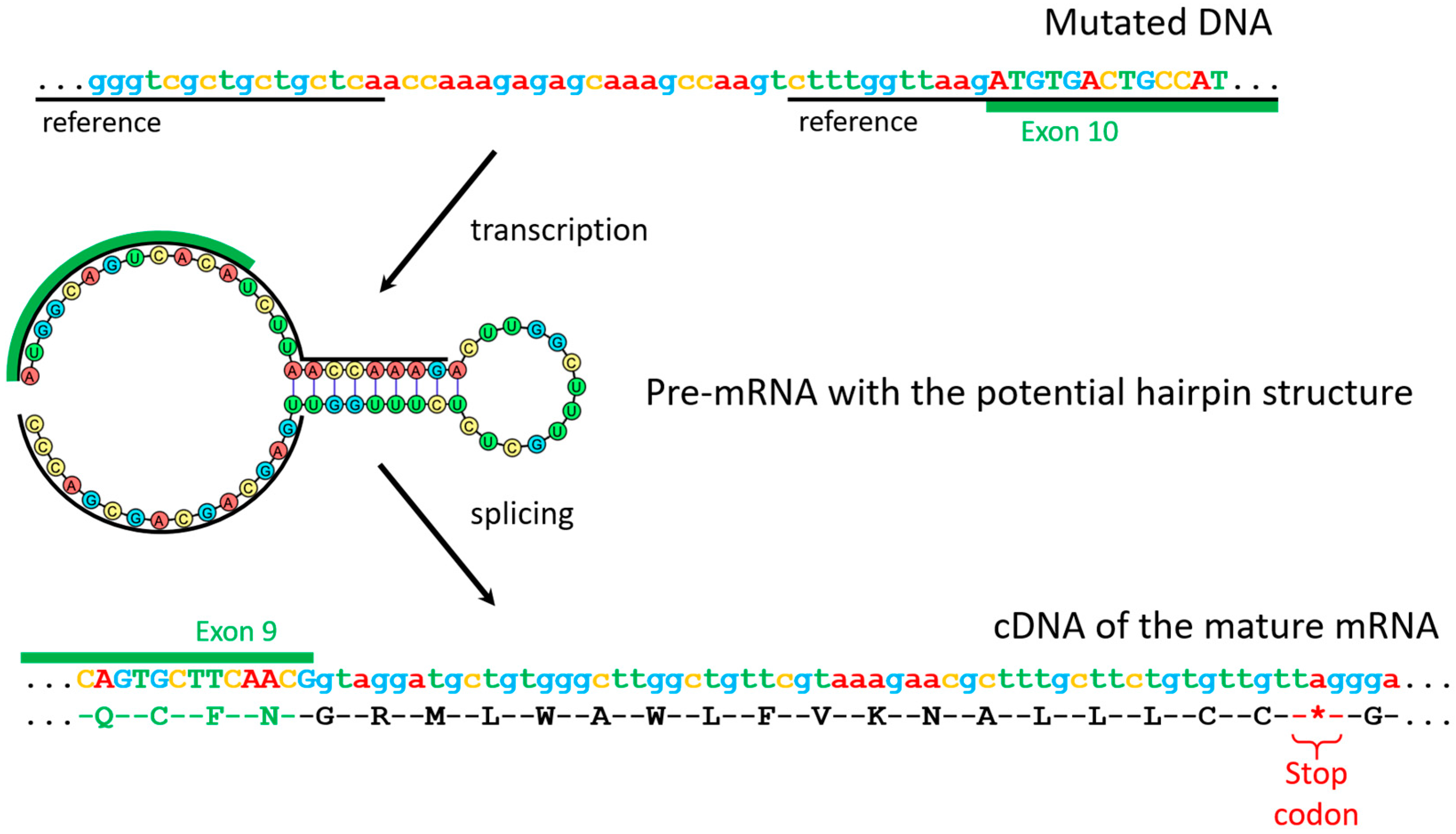
3.6. Genetic Verification of Pseudohypoparathyroidism by GNAS Sequencing
4. Discussion
- Multihormonal resistance, including PTH, TSH, LH, FSH, somatotropin-releasing hormone, etc. [14].
- Phenotypic manifestations of AHO, including:
- Roundness of the face;
- Obesity, often ahead of other endocrine disorders [15];
- Shortening of the IV and/or V metacarpal and metatarsal bones, and the distal phalanx of the big toe. Brachydactyly is formed in the process of accelerated closure of the growth zones and is caused by impaired signal transduction from PTH1R in chondrocytes due to a Gαs defect. In patients with GNAS inactivation, brachydactyly is not present at birth but develops over time. Shortening of the metacarpal bones leads to pits in the metacarpophalangeal joints of four of the five fingers, which is manifested by the classic sign of “knuckle, knuckle, dimple, dimple” when the hand is clenched into a fist;
- Ectopic ossification of soft tissues. The main cause is the deficiency of Gαs in mesenchymal stem cells, which contributes to their differentiation into osteoblasts in extra-osseous areas—subcutaneous tissue and dermis, followed by the formation of new bone tissue [16];
- Decrease in mental abilities of varying severity [17]. Impairment of cognitive functions is detected in about half of patients with PHP Ia. The cause, apparently, is a Gαs defect, but not chronic hypocalcemia, since mental abilities are preserved in patients with other forms of PHP and a low level of blood calcium.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Romanet, P.; Galluso, J.; Kamenicky, P.; Hage, M.; Theodoropoulou, M.; Roche, C.; Graillon, T.; Etchevers, H.; De Murat, D.; Mougel, G.; et al. Somatotroph Tumors and the Epigenetic Status of the GNAS Locus. Int. J. Mol. Sci. 2021, 22, 7570. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, P.; Teisinger, J.; Novotny, J.; Bourová, L.; Drmota, T.; Hejnova, L.; Moravcová, Z.; Lisý, V.; Rudajev, V.; Stöhr, J.; et al. Biochemistry of transmembrane signaling mediated by trimeric G proteins. Physiol. Res. 2004, 53 (Suppl. S1), S141–S152. [Google Scholar] [PubMed]
- Weinstein, L.S.; Chen, M.; Liu, J. Gs(alpha) Mutations and Imprinting Defects in Human Disease. Ann. N. Y. Acad. Sci. 2002, 968, 173–197. Available online: https://pubmed.ncbi.nlm.nih.gov/12119276/ (accessed on 16 December 2021). [CrossRef] [PubMed]
- Hayward, B.E.; Moran, V.; Strain, L.; Bonthron, D.T. Bidirectional Imprinting of a Single Gene: GNAS1 Encodes Maternally, Paternally, and Biallelically Derived Proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 15475–15480. Available online: https://pubmed.ncbi.nlm.nih.gov/9860993/ (accessed on 16 December 2021). [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yu, S.; Litman, D.; Chen, W.; Weinstein, L.S. Identification of a Methylation Imprint Mark within the Mouse Gnas Locus. Mol. Cell Biol. 2000, 20, 5808–5817. Available online: https://pubmed.ncbi.nlm.nih.gov/10913164/ (accessed on 16 December 2021). [CrossRef] [Green Version]
- Li, X.; Murray, F.; Koide, N.; Goldstone, J.; Dann, S.M.; Chen, J.; Bertin, S.; Fu, G.; Weinstein, L.S.; Chen, M.; et al. Divergent Requirement for Gαs and cAMP in the Differentiation and Inflammatory Profile of Distinct Mouse Th Subsets. J. Clin. Investig. 2012, 122, 963–973. Available online: https://pubmed.ncbi.nlm.nih.gov/22326954/ (accessed on 16 December 2021). [CrossRef] [Green Version]
- Chen, M.; Shrestha, Y.B.; Podyma, B.; Cui, Z.; Naglieri, B.; Sun, H.; Ho, T.; Wilson, E.A.; Li, Y.-Q.; Gavrilova, O.; et al. Gsα Deficiency in the Dorsomedial Hypothalamus Underlies Obesity Associated with Gsα Mutations. J. Clin. Investig. 2017, 127, 500–510. Available online: https://pubmed.ncbi.nlm.nih.gov/27991864/ (accessed on 16 December 2021). [CrossRef] [Green Version]
- Klenke, S.; Siffert, W.; Frey, U.H. A Novel Aspect of GNAS Imprinting: Higher Maternal Expression of Gαs in Human Lymphoblasts, Peripheral Blood Mononuclear Cells, Mammary Adipose Tissue, and Heart. Mol. Cell Endocrinol. 2011, 341, 63–70. Available online: https://pubmed.ncbi.nlm.nih.gov/21664251/ (accessed on 16 December 2021). [CrossRef] [Green Version]
- Mantovani, G. Clinical Review: Pseudohypoparathyroidism: Diagnosis and Treatment. J. Clin. Endocrinol. Metab. 2011, 96, 3020–3030. Available online: https://pubmed.ncbi.nlm.nih.gov/21816789/ (accessed on 16 December 2021). [CrossRef] [Green Version]
- Mantovani, G.; Lecumberri, B.; Bastepe, M.; Monk, D.; de Sanctis, L.; Thiele, S.; Usardi, A.; Ahmed, F.; Bufo, R.; Choplin, T.; et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: First international Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 476–500. [Google Scholar] [CrossRef]
- Zhou, Q.; Liang, B.; Fu, Q.-X.; Liu, H.; Zou, C.-C. Different AHO phenotype in a Chinese family with a novel GNAS missense variant: A case report. Italy J. Pediatr. 2022, 48, 123. [Google Scholar] [CrossRef]
- Pereda, A.; Elli, F.M.; Thiele, S.; de Sanctis, L.; Rothenbuhler, A.; Hanna, P.; Francou, B.; Ertl, D.A.; de Nanclares, G.P.; Linglart, A.; et al. Inactivating PTH/PTHrP signaling disorders (iPPSDs): Evaluation of the new classification in a multicenter large series of 544 molecularly characterized patients. Eur. J. Endocrinol. 2021, 184, 311–320. [Google Scholar] [CrossRef]
- Thiele, S.; Mantovani, G.; Barlier, A.; Boldrin, V.; Bordogna, P.; De Sanctis, L.; Elli, F.M.; Freson, K.; Garin, I.; Grybek, V.; et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur. J. Endocrinol. 2016, 175, P1–P17. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, G.; Francesca Marta, E. Multiple hormone resistance and alterations of G-protein-coupled receptors signaling. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Kayemba-Kay’s, S.; Tripon, C.; Heron, A.; Hindmarsh, P. Pseudohypoparathyroidism Type 1A-Subclinical Hypothyroidism and Rapid Weight Gain as Early Clinical Signs: A Clinical Review of 10 Cases. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 432. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Xu, M.; Russell, E.; Richardson, A.; Kaplan, J.; Billings, P.C.; Kaplan, F.S.; Shore, E.M. Heterozygous inactivation of Gnas in adipose-derived mesenchymal progenitor cells enhances osteoblast differentiation and promotes heterotopic ossification. J. Bone Min. Res. 2011, 26, 2647–2655. [Google Scholar] [CrossRef] [Green Version]
- Mouallem, M.; Shaharabany, M.; Weintrob, N.; Shalitin, S.; Nagelberg, N.; Shapira, H.; Zadik, Z.; Farfel, Z. Cognitive impairment is prevalent in pseudohypoparathyroidism type Ia, but not in pseudopseudohypoparathyroidism: Possible cerebral imprinting of Gsalpha. Clin. Endocrinol. 2008, 68, 233–239. [Google Scholar] [CrossRef]
- Levine, M.A.; Germain-Lee, E.; De Beur, S.J. Genetic basis for resistance to parathyroid hormone. Horm Res. 2003, 60 (Suppl. S3), 87–95. [Google Scholar] [CrossRef]
- Shoback, D.M.; Bilezikian, J.P.; Costa, A.G.; Dempster, D.; Dralle, H.; Khan, A.A.; Peacock, M.; Raffaelli, M.; Silva, B.C.; Thakker, R.; et al. Presentation of Hypoparathyroidism: Etiologies and Clinical Features. J. Clin. Endocrinol. Metab. 2016, 101, 2300–2312. [Google Scholar] [CrossRef]
- Subbiah, S.; Natarajan, V.; Bhagadurshah, R.R. Fahr’s Disease and Hypoparathyroidism—A Missing Link. Neurol. India 2022, 70, 1159–1161. [Google Scholar]
- Rodriguez, H.J.; Villarreal, H.; Klahr, S.; Slatopolsky, E. Pseudohypoparathyroidism type II: Restoration of normal renal responsiveness to parathyroid hormone by calcium administration. J. Clin. Endocrinol. Metab. 1974, 39, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Lemos, M.C.; Thakker, R.V. GNAS mutations in Pseudohypoparathyroidism type 1a and related disorders. Hum. Mutat. 2015, 36, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, K.S.; Chang, C.C.; Wu, D.J.; Huang, T.S.; Tsai, I.H.; Chen, F.W. Deficient erythrocyte membrane Gs alpha activity and resistance to trophic hormones of multiple endocrine organs in two cases of pseudohypoparathyroidism. Taiwan Yi Xue Hui Za Zhi 1989, 88, 450–455. [Google Scholar]
- Ridderskamp, P.; Schlaghecke, R. Pseudohypoparathyroidism and adrenal cortex insufficiency. A case of multiple endocrinopathy due to peripheral hormone resistance. Klin. Wochenschr. 1990, 68, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, S.K.; Sangla, K.S. A sporadic case of pseudohypoparathyroidism type 1 and idiopathic primary adrenal insufficiency associated with a novel mutation in the GNAS1 gene. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2014, 20, e202–e206. [Google Scholar] [CrossRef]
- Bollerslev, J.; Rejnmark, L.; Marcocci, C.; Shoback, D.M.; Sitges-Serra, A.; Van Biesen, W.; Dekkers, O.M.; European Society of Endocrinology. European Society of Endocrinology Clinical Guideline: Treatment of chronic hypoparathyroidism in adults. Eur. J. Endocrinol. 2015, 173, G1–G20. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.W.; Nebesio, T.D.; DiMeglio, L.A.; Eugster, E.A.; Imel, E.A. Prevalence of Nephrocalcinosis in Pseudohypoparathyroidism: Is Screening Necessary? J. Pediatr. 2018, 199, 263–266. [Google Scholar] [CrossRef]
- Wu, Y.L.; Hwang, D.Y.; Hsiao, H.P.; Ting, W.H.; Huang, C.Y.; Tsai, W.Y.; Chen, H.-C.; Chao, M.-C.; Lo, F.-S.; Tsai, J.-D.; et al. Mutations in Pseudohypoparathyroidism 1a and Pseudopseudohypoparathyroidism in Ethnic Chinese. PLoS ONE 2014, 9, e90640. Available online: https://pubmed.ncbi.nlm.nih.gov/24651309/ (accessed on 16 December 2021).
- Ham, H.J.; Baek, K.H.; Lee, J.Y.; Kim, S.Y.; Mo, E.Y.; Kim, E.S.; Han, J.H.; Moon, S.-D. Analysis of Aberrantly Spliced Transcripts of a Novel De Novo GNAS Mutant in a Male with Albright Hereditary Osteodystrophy and PHP1A. Horm. Metab. Res. 2015, 47, 585–590. Available online: https://pubmed.ncbi.nlm.nih.gov/25502941/ (accessed on 16 December 2021). [CrossRef]
- Fischer, J.A.; Egert, F.; Werder, E.; Born, W. An Inherited Mutation Associated with Functional Deficiency of the α-Subunit of the Guanine Nucleotide-Binding Protein Gs in Pseudo- and Pseudopseudohypoparathyroidism1. J. Clin. Endocrinol. Metab. 1998, 3, 935–938. [Google Scholar]
- Rickard, S.J.; Wilson, L.C. Analysis of GNAS1 and overlapping transcripts identifies the parental origin of mutations in patients with sporadic Albright hereditary osteodystrophy and reveals a model system in which to observe the effects of splicing mutations on translated and untranslated messenger RNA. Am. J. Hum. Genet. 2003, 72, 961–974. [Google Scholar]
- Lietman, S.A. Preimplantation genetic diagnosis for hereditary endocrine disease. Endocr. Pract. 2011, 17, 28–32. [Google Scholar] [CrossRef]
- Chang, G.; Li, Q.; Li, N.; Li, G.; Li, J.; Ding, Y.; Huang, X.; Shen, Y.; Wang, J.; Wang, X. Evaluating the variety of GNAS inactivation disorders and their clinical manifestations in 11 Chinese children. BMC Endocr. Disord. 2022, 22, 70. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Date | Ca total, mmol/L (2.15–2.55) | Ca ionized, mmol/L (1.03–1.29) | P, mmol/L (0.74–1.52) | PTH, pg/mL (15–65) | Na, mmol/L (136–145) | TSH, mIU/L (0.25–3.5) | AP, IU/L (40–150) | Medication |
|---|---|---|---|---|---|---|---|---|
| 2001 | - | - | - | - | - | 6.7 | - | Cholecalciferol 5000 IU/day Levothyroxine 12.5 mcg/day |
| 2003 | - | - | - | - | - | 10.0 | - | Alfacalcidol 1.2 mcg/day Levothyroxine 75 mcg/day |
| May 2004 | 1.9 | - | 2.4 | - | - | 4.3 | 909 | Previous therapy |
| August 2004 | 1.9 | 2.0 | - | - | - | 803 | Previous therapy | |
| December 2004 | 1.9 | 1.17 | - | - | - | - | Previous therapy | |
| 2005 | - | - | - | - | - | 7.5 | - | Alfacalcidol 1.2 mcg/day Levothyroxine 100 mcg/day Desmopressin 0.2 mg twice daily |
| 2008 | 1.71 | 0.88 | 2.97 | 127 | 5.29 | - | Cholecalciferol 25,000 IU/day Levothyroxine 125 mcg/day Desmopressin (dose data are not available) | |
| 2010 | - | - | - | - | - | 15.5 | - | Cholecalciferol 25,000 IU/day Levothyroxine 150 mcg/day Desmopressin (dose data are not available |
| 2014 | 2.12 | 1.14 | 3.1 | 236.5 | 135.4 | 2.72 | 521 | Cholecalciferol 50,000 IU/day Levothyroxine 150 mcg/day Desmopressin (dose data are not available |
| February 2021 | - | - | - | - | - | - | - | Cholecalciferol 50,000 IU/day Levothyroxine 150 mcg/day Desmopressin 120 mcg/day |
| Parameter | Value | References | Parameter | Value | References |
|---|---|---|---|---|---|
| Morning ACTH | 103.9 pg/mL | 7.2–63.3 | Albumen | 49 g/L | 35–50 |
| Evening ACTH | 26.76 pg/mL | 2–25.5 | Sodium | 139 mmol/L | 136–145 |
| TSH | 2.145 mIU/L | 0.25–3.5 | Chlorides | 101 mmol/L | 98–107 |
| FSH | 3.54 U/L | 1.6–9.7 | Potassium | 3.9 mmol/L | 3.5–5.1 |
| LH | 10.1 U/L | 2.5–11 | Glucose | 4.69 mmol/L | 3.1–6.1 |
| Testosterone | 9.59 nmol/L | 11–28.2 | HbA1c | 5% | 4–6 |
| SHBG | 23.99 nmol/L | 18.3–54.1 | Creatinine | 65.8 mcmol/l | 63–110 |
| IGF-1 | 301.7 ng/mL | 119–511 | Phosphorus | 1.33 mmol/L | 0.74–1.52 |
| STH | 0.093 ng/mL | 0.02–1.23 | AP | 68 U/L | 40–150 |
| Blood cortisol in the morning | 487.5 nmol/L | 171–536 | Total cholesterol | 4.52 mmol/L | 3.3–5.2 |
| Evening saliva cortisol | 10.11 nmol/L | 0.5–9.65 | LDL | 2.8 mmol/L | 1.1–3 |
| Plasma osmolality | 287 mOsm/kg | 280–300 | HDL | 1.278 mmol/L | 0.9–2.6 |
| Urine osmolality | 703 mOsm/kg | 300–1200 | Triglycerides | 1.06 mmol/L | 0.1–1.7 |
| PSA (total) | 0.25 ng/mL | 0–4 | ALT | 14 U/L | 0–55 |
| Magnesium | 0.84 mmol/L | 0.7–1.05 | AST | 17 U/L | 5–34 |
| Parameter | Value, ng/mL | References |
|---|---|---|
| 24,25-(OH)2-D3 | 22.6 | 0.5–5.6 |
| 25-(OH)-D3/24,25-(OH)2-D3 | 12.654867 | 7–25 |
| 25-(OH)-D3 | 275 | 20–60 |
| 25-(OH)-D2 | 0.01 | 20–60 |
| Total 25-(OH)-D | 275.01 | 20–60 |
| 3-epi-25-(OH)-D3 | 22.7 | 1–10 |
| Mutation | Location of Splicing Mutation | Effect on Pre-mRNA Splicing | Commentary | Reference |
|---|---|---|---|---|
| c.119_139 + 17del | Exon 1/intron 1 boundary | Partial deletion (21bp) of exon 1 | The 38 bp deletion at the exon 1/intron 1 boundary comprising 21 nucleotides of the 3’-end of exon 1 and 17 nucleotides of intron 1 in the mutated allele. This eliminates the donor splice site of exon 1, giving rise to a transcript that includes intron 1. As a result, termination of translation is predicted to occur within intron 1, leading to the incorporation of at least 116 alternative amino acids into a protein product of the mutated Gsα gene. | [30] |
| c.313-11A > G | Intronic | Retention of intron 4 | The inclusion of intron 4 in the Gαs transcript is predicted to result in premature truncation 21 codons downstream. | [31] |
| c.312+5G > A | Intronic | Exon 4 skipping | This change is predicted to result in aberrant splicing with removal of exon 4. | [32] |
| c.212 + 3_212 + 6delAAGT | Intronic | Cryptic exon inclusion | Predicted to disrupt the highly conserved 5’ splice site sequence in intron 2 and activate a cryptic splice site located 28 bp downstream of the intron. | [33] |
| c.719-29_719-13delinsACCAAAGAGAGCAAAGCCAAG | Intronic | Cryptic exon inclusion | Sequencing revealed that, in addition to the exons of the gene, it includes a 108-nucleotide sequence from the mutant ninth intron that contains a stop codon. | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorbacheva, A.; Pogoda, T.; Bogdanov, V.; Zakharova, V.; Salimkhanov, R.; Eremkina, A.; Melnichenko, G.; Mokrysheva, N. A Novel GNAS Mutation in a Patient with Ia Pseudohypoparathyroidism (iPPSD2) Phenotype. Genes 2023, 14, 324. https://doi.org/10.3390/genes14020324
Gorbacheva A, Pogoda T, Bogdanov V, Zakharova V, Salimkhanov R, Eremkina A, Melnichenko G, Mokrysheva N. A Novel GNAS Mutation in a Patient with Ia Pseudohypoparathyroidism (iPPSD2) Phenotype. Genes. 2023; 14(2):324. https://doi.org/10.3390/genes14020324
Chicago/Turabian StyleGorbacheva, Anna, Tatyana Pogoda, Viktor Bogdanov, Victoriya Zakharova, Rustam Salimkhanov, Anna Eremkina, Galina Melnichenko, and Natalia Mokrysheva. 2023. "A Novel GNAS Mutation in a Patient with Ia Pseudohypoparathyroidism (iPPSD2) Phenotype" Genes 14, no. 2: 324. https://doi.org/10.3390/genes14020324